
Laboratory analysis
The aim of laboratory analyses is to obtain accurate and precise data for your water quality monitoring program in a safe environment.
Check with relevant lo cal authorities in your jurisdiction who might have established procedures for laboratory analyses.
A checklist of important considerations is given in Box 1. Download our laboratory request form example.
Box 1 Checklist for undertaking laboratory analyses
- Have the analytes been clearly stated?
- Have appropriate analytical methods been identified?
– Will analytical methods cover the range of concentrations expected?
– Will analytical methods detect the minimum concentration of interest?
– Will the analytical methods have sufficient accuracy and precision?
– Will the substances be processed within the samples’ storage life? - Does the laboratory have the appropriate equipment to undertake the analytical method chosen?
- Are laboratory facilities (water supply, air supply, environment) suitable for the planned analyses?
- Do the laboratory staff have the expertise, training and competence to undertake the planned analyses?
- Has a laboratory data management system been established? If so, does the laboratory data management system:
– track samples and data (chain of custody)?
– have written data entry protocols to ensure correct entry of data?
– enable associated data to be retrieved, e.g. nutrient concentrations and flows to calculate nutrient loads?
– have validation procedures to check accuracy of data?
– have appropriate storage and retrieval facilities to prevent loss of data and enable retrieval (for at least three years) based on current and unexpected information needs? - Are procedures in place to ensure information has reached the user?
- From the documentation, can you see:
– how results were obtained?
– that samples had unique identification?
– who the analyst was?
– what test equipment was used?
– the original observations and calculations?
– how data transfers occurred?
– how standards were prepared?
– the certified calibration solutions used, their stability and storage? - Has a laboratory quality assurance plan been developed?
- Does the laboratory quality assurance plan specifically cover:
– operating principles?
– training requirements for staff?
– preventative maintenance of laboratory infrastructure and equipment?
– the requirements of the laboratory data management system?
– procedures for when and how corrective actions are to be taken?
– allocation of responsibility to laboratory staff?
– all documentation to maintain quality at specified levels?
– quality control procedures to minimise analysis errors?
– quality assessment procedures to determine quality of data? - Are all protocols for preparing and analysing samples written and validated?
- Are standard methods being used? If variations of standard methods or non-standard procedures are used, procedures must be technically justified, with documentation of the effects of changes.
- Have analytical methods’ accuracy, bias and precision been established by:
– analysis of standards?
– independent methods?
– recovery of known additions?
– analysis of calibration check standards?
– analysis of reagent blanks?
– analysis of replicate samples? - Do operation procedures specify instrument optimisation and calibration methods, and do these cover:
– preventative maintenance?
– specific optimisation procedures?
– resolution checks?
– daily calibration procedures?
– daily performance checks? - With respect to laboratory QA/QC:
– Have control charts for laboratory control standards, calibration check standards, reagent blanks and replicate analyses been established?
– Does the laboratory participate in proficiency testing programs?
– Does the laboratory conduct unscheduled performance audits in which deviations from standard operating procedures (protocols) are identified and corrective action taken? - Have all reasonable practical steps been taken to protect the health and safety of laboratory staff?
– Have hazards been identified?
– Have laboratory staff been educated about hazards?
– Have risk minimisation plans been prepared?
– Have staff been trained to ensure safe work practices?
– Are staff appropriately supervised?
– Are staff insured?
Analytes
The particular substances to be analysed (indicators, or analytes for laboratory purposes) are the focus of the monitoring program at Step 3 of the Water Quality Management Framework. They may have been identified in generic terms during the study design, but now the individual compounds need to be decided on, and possible methods of determination need to be considered before planning the laboratory program.
The analytes will determine many of the decisions involved in laboratory analyses: for example, how to obtain good quality data (method and equipment), how to protect the health and safety of workers, and how much this stage of the monitoring program will cost.
Choice of analytical methods
The selection of an analytical method for waters, sediments or biota will largely depend on the information and management needs of those undertaking the investigation, and on the analytes themselves. However, limitations such as the financial resources available, laboratory resources, speed of analyses required, matrix type and contamination potential, are also important factors.
Your choice of an appropriate analytical method is based on 4 considerations:
- range of analyte concentrations to be determined — detection limits are method-specific and the lowest concentration of interest will need to be specified
- accuracy and precision needed — all results are only estimates of the true value, and the greater the accuracy and precision you require, the greater the analytical complexity and cost
- maximum period between sampling and analysis — on-the-spot field analysis may be required, depending on the use to be made of the data
- familiarity with method and availability of analytical instrumentation.
Appropriate procedures for both biological and chemical analyses can be found by reference to accepted published procedures, such as APHA (2012) or other approved sampling and analysis methods (e.g. DEC 2004, EPAV 2009). The US Environmental Protection Authority has published methods that cover water, sediment and biological analyses. Detailed reviews or descriptions of methods are available in standard texts. Methods for marine water analysis are found in Grasshoff et al. (2007).
Analytical methods and associated references for biological, chemical and physical parameters are summarised in Tables 1 to 3. Methods for toxicity testing are not included but have been summarised in Section 3.5 of the ANZECC & ARMCANZ (2000) guidelines.
Standard methods are updated regularly but with the rapid pace of research, many good non-standard methods are available that have not yet been included in standard methods compilations. Their use is acceptable, provided that justification for their choice is given and that their performance can be demonstrated through the analysis of standard reference materials or other quality control procedures.
Aspects or details of both standard and non-standard procedures may require evaluation or modification for use in Australian or New Zealand conditions. Some of these modifications are well documented but others either are not recorded or are recorded in commercially available methods.
| Analyte | Methods | References* |
|---|---|---|
| Bioaccumulation | Standard analytical procedures | APHA 2012 |
| Coliform bacteria | Enumeration | APHA 2012 |
| Toxicity testing | – | ASTM 2013 |
* for APHA (2012) use this or the most current edition
| Analyte | Methods | References* |
|---|---|---|
| Ammonia | Ammonia electrode, titrimetry, colorimetry | APHA 2012, USEPA 1983 |
| Biological oxygen demand | Incubation | APHA 2012, USEPA 1983 |
| Carbamate pesticides | High-performance liquid chromatography (HPLC) | USEPA 1999 |
| Carbonate, bicarbonate | Titrimetry | APHA 2012 |
| Chemical oxygen demand | Reflux, titrimetry, colorimetry | APHA 2012, USEPA 1983 |
| Chloride | Potentiometry, titrimetry | APHA 2012 |
| Chlorinated phenoxy acid herbicides | Gas chromatophraphy (GC) | APHA 2012 |
| Chlorine | Iodometry, amperometry | APHA 2012 |
| Chlorophyll | Fluorimetry, spectrophotometry | APHA 2012 |
| Cyanide | Titrimetry, colorimetry, ion selective electrode | APHA 2012 |
| Dioxins | Gas chromatography–mass spectrometry (GC–MS) | APHA 2012 |
| Dissolved oxygen | Iodometry, oxygen electrode, Winkler method | APHA 2012, USEPA 1983 |
| Hardness | Titrimetry | APHA 2012 |
| Metals | Inductively coupled plasma atomic emission spectroscopy (ICPAES), inductively coupled plasma mass spectrometry (ICPMS), atomic absorption spectroscopy (AAS), plus specialist methods for Al, Hg, As, Se, Cr(VI), speciation | APHA 1998, USEPA 1983, 1994, USEPA 2002a (Hg), USEPA 1996 (As) |
| Oil and grease | Assorted | APHA 2012 |
| Organic carbon | Combustion infrared, persulfate UV | APHA 2012, USEPA 1983 |
| Organochlorine compounds | GC all | APHA 2012 |
| Organophosphate pesticides | GC–MS | APHA 2012 |
| pH, alkalinity, acidity | Electrometry, titration | APHA 2012, USEPA 1983 |
| Phenols | Assorted | APHA 2012 |
| Phosphorus | Colorimetry | APHA 2012 |
| Polycyclic aromatic hydrocarbons (PAHs) | GC, GC–MS | APHA 2012 |
| Nitrate, nitrite | Colorimetry, titrimetry | APHA 2012, USEPA 1983, Grasshoff et al. 2007 |
| Radioactivity | Counting | – |
| Salinity | Electrical conductivity, density, sensors, titration | APHA 1998, Grasshoff et al. 2007, Parsons et al. 1985 |
| Silica | AAS, colorimetry, ICPAES | APHA 2012 |
| Surfactants | Spectrophotometry, etc. | APHA 2012 |
| Sulfur compounds | Assorted | APHA 2012 |
| Total Kjeldahl nitrogen | Colorimetry, potentiometry | APHA 2012, USEPA 1983 |
* for APHA (2012) and USEPA volumes, use these or the most current editions
| Analyte | Methods | References |
|---|---|---|
| Clarity | Secchi disk | APHA 2012 |
| Colour | Colorimetry | APHA 2012, USEPA 1983 |
| Conductivity | Instrumental | APHA 2012 |
| Depth | Acoustic Doppler current profiler, depth sounder | RD Instruments 1989, EPA 1992 |
| Flow rates | Acoustic Doppler current profiler, assorted methods | USEPA 1982, RD Instruments 1989 |
| Gross contamination | Floatables, solvent-soluble floatable oil and grease | APHA 2012 |
| Suspended solids | Gravimetry | APHA 2012, USEPA 1983, AS 1990 |
| Temperature | Thermometer, electronic data logger, thermistor | – |
| Turbidity | Nephelometry, light scattering | APHA 2012, USEPA 1983 |
* for APHA (2012), USEPA volumes and AS (1990), use these or the most current editions
We provide considerable information on the modes of action of the various parameters, their effects on human health, and default guideline values (DGVs) on tolerable concentration limits in the Toxicant DGVs technical briefs.
You need to ensure that environmentally responsible methods are chosen whenever possible. Waste disposal, in particular, must be considered when deciding on a suitable method.
It may not be necessary to use an elaborate and expensive method if a more up-to-date, cost-effective and less environmentally damaging technique (e.g. a field testing procedure) may provide information at the required level of accuracy.
Before undertaking analyses, your monitoring team and the end-users of the data should confirm that the chosen laboratory has the appropriate equipment, expertise and experience to undertake the analytical method chosen, as well as an adequate quality assurance/quality control (QA/QC) program.
If your monitoring team plans to send samples to external laboratories, we recommend using laboratories accredited by the National Association of Testing Authorities (NATA) wherever possible. Accreditation guarantees appropriate standards for laboratory organisation and QA/QC but not necessarily accurate results.
Results reported in a variety of different units (e.g. g/L, ppm, molarity, number of organisms) can cause confusion and wasted effort when different sources are combined or compared. We recommend that you adopt a system of consistent units, such as those in the Water Quality Guidelines (mass/L or mass/kg, as appropriate).
Data management
When samples are delivered to the laboratory for analysis, it is essential that the laboratory staff log the samples into the laboratory register or record system, and give each a unique identification code. This becomes part of the chain of custody of the sample.
The record system needs to:
- provide a traceable pathway covering all activities from receipt of samples to disposal
- allow retrieval, for a period of at least 3 years, of all original test data within the terms of registration.
Data storage
System design considerations
Water quality data are expensive and time-consuming to collect so they must be made as useable, useful and retrievable as possible through careful systematic storage.
The sheer volume of data accumulated by any monitoring program after just a few years of operation dictates that computer-based data management systems must be used to store and manage data.
A data management system should have:
- reliable procedures for recording results of analysis or field observations
- procedures for systematic screening and validation of data
- secure storage of information
- a simple retrieval system
- simple means of analysing data
- flexibility to accommodate additional information (e.g. analytes, sites).
Water quality database designers must consider user needs, including:
- scope of the data to be stored — sources, numbers of samples, sample identifiers (e.g. identification number, type, site, time or date of collection), number of database fields, descriptive notes, confidence categories, analytes, analysis types, number of records
- issues caused by multiple sources of data — validation and standardisation procedures, transfer formats, confidence ratings
- QA/QC (risk/confidence levels) — analytical precision, validation procedures
- linkage to flow or tides
- documentation — standard methods of analysis, validation procedures, codes
- how the data will be accessed by users — online at the same time as it is generated (real-time), online retrieval, data retrieval request-based system, categories of data (macro design)
- types of data analysis support required — statistical, graphical, trend analysis, regression analysis.
ANZLIC — the Spatial Information Council has published AS/NZS ISO 19115.1:2015 Metadata as a standard for developing metadata for geographic information.
Data tracking
If data are to be the basis for legal proceedings at some time in the future, a chain of custody is particularly important. In this context, the laboratory may be asked these questions:
- How was each sample labelled to ensure no possibility of mix up or substitution?
- How were the data identified to ensure no mix up or substitution?
Your laboratory record system will ensure the integrity of the sample from collection to final analysis with respect to the variables of interest. If all data have unique identification codes, then chain of custody documentation ensures that these questions can be answered.
Screening and verification
Data entry protocols must be developed to ensure that the entry of data is accurate. Data from instruments should be electronically transferred to the database where possible to prevent transcription errors.
Harmonisation of data
Harmonised data are data that can be used or compared with data from other datasets in comparable units of measurement or time frames.
For example, if nutrient loads are to be calculated, then concentration data and flow data must be collected at the same location at the same time.
To ensure that data are harmonised and can be usefully compared, your monitoring team must consider making additional measurements.
Retrieval and sharing of data in databases
Many different databases have been developed — often associated with the operation systems of particular authorities (water supply, waste water management, storage management). Some of these systems have been costly to update for use with new computer technologies, and have been incompatible with other databases, resulting in difficulties in transferring data although data interchange has been greatly facilitated in recent years by more pervasive use of SQL databases.
Substantial growth in electronic transfer and online access to data has encouraged standardisation of databases. The choice of a particular database depends on the types and intended uses of the data, and the type and compatibility of the computer hardware and software. The data should be available and able to be shared with other databases for years to come, through platforms like data.gov.au.
Laboratory data reporting
Where separate laboratory reports are provided as part of a monitoring program, these should include:
- laboratory name and address
- tabulation of samples and analysis data
- identification of the analytical methods used
- date of analysis and name of technician or chemist
- quality assurance statement.
These details are sometimes reproduced in full, for all relevant samples, in the appendixes of a primary report; otherwise the most appropriate data are abstracted and listed in the body of the report.
QA/QC in laboratory analyses
The objectives of a QA/QC program in a laboratory are to:
- minimise errors that can occur during subsampling and analytical measurement
- produce data that are accurate, reliable and acceptable to the data user.
QA/QC procedures are designed to prevent, detect and correct problems in the measurement process and to characterise errors statistically, through quality control samples and various checks.
Here we consider what is expected of the laboratory undertaking the analyses. End users of the data should receive evidence that data meet their QA/QC requirements.
Traceability of results
Traceability of analytical results from the laboratory report back to the original sample is an essential component of good laboratory practice and a prerequisite for accreditation of analytical laboratories.
Apart from the chain of custody details for each sample, for each analysis the laboratory record system must include:
- identity of the sample analysed
- identity of analyst
- name of equipment used
- original data and calculations
- identification of manual data transfers
- documentation of standards preparation
- use of certified calibration solutions.
Laboratory facilities
Laboratory facilities and environment must be clean, with appropriate consideration of work health and safety issues.
Check for airborne contamination regularly; it can enter through air conditioning systems or be generated internally from users of the laboratory.
Deionised water is the most extensively used reagent in the laboratory and it must be maintained at the appropriate standard required to conduct analyses. The electrical conductivity of the deionised water should be monitored continuously or on a daily basis, with the water being checked regularly for trace metals and organic compounds.
Analytical equipment
All equipment and laboratory instruments should be kept clean and in good working order, with up-to-date records of calibrations and preventative maintenance. Record any repairs to equipment and instruments, as well as details of any incidents that may affect the reliability of the equipment.
Human resources
All staff undertaking analyses must be technically competent, skilled in the particular techniques being used and have a professional attitude towards their work. Train staff in all aspects of the analyses being undertaken.
Before analysts are permitted to do reportable work, they must demonstrate their competence to undertake laboratory measurements. As a minimum, they should be able to show they can adhere to a written protocol and that their laboratory practices do not contaminate samples. They should demonstrate their ability to work safely in the laboratory and to use the prescribed methods to obtain reproducible data that are of acceptable accuracy and precision.
QA/QC in analytical protocols
Laboratories undertaking analyses must fully document the methods used. Methods must be described in sufficient detail so that an experienced analyst unfamiliar with a method can reproduce it and obtain acceptable results.
Laboratory staff should be aware that it is important to adhere strictly to analytical protocols. They should appreciate the critical relevance of rigorous quality control and assurance in the laboratory.
Proper laboratory practice is codified in the requirements of registration authorities, such as NATA. Laboratories holding registration from NATA and similar organisations will be familiar with the effort required to achieve and maintain a facility with creditable performance standards.
All laboratories must have a formal system of periodically reviewing the technical suitability of analytical methods. If standard methods are used, it is not enough to quote the standard method; any variation of the standard method must be technically justified and supported by a documented study on the effects of the changes.
Measurement errors can be categorised as:
- random errors — affect the precision of the results (degree to which data generated from repetitive measurements of a sample or samples will differ from one another). Statistically this is expressed as the standard deviation (SD) for the replicate measurements of an individual sample and the standard error for replicate measurements of a number of samples. It may also be given as the coefficient of variation (SD divided by the mean, expressed as a percentage). This is often referred to as repeatability or reproducibility. Sources of random error include spurious contamination, electronic noise and uncertainties in pipetting and weighing.
- systematic errors or biases — result in differences between the mean and the true value of the analyte of concern (accuracy). Systematic errors can only be established by comparing the results obtained against the known or consensus values. Sources of systematic error include reagent contamination, instrument calibration and method interferences.
The principal indicators of data quality are:
- bias — a measurement of systematic error that can be attributed either to the method or to the laboratory’s use of the method
- precision — amount of agreement between multiple analyses of a given sample (APHA 2012).
When combined, bias and precision are expressed as accuracy (nearness of the mean of a set of measurements to the ‘true’ value) (APHA 2012). The data can be referred to as being ‘accurate’ when the bias is low and the precision is high (Figure 2).
Figure 2 Illustration of accuracy in terms of bias and precision (modified from APHA 2012)
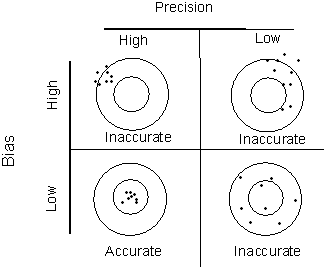
Quality assessment is the process of using standard techniques to assess the accuracy and precision of measurement processes and for detecting contamination.
For example, the accuracy of analytical methods can be established by:
- analysis of reference materials
- inter-laboratory collaborative testing programs (in which it is assumed that the consensus values for analytes are true)
- performance audits
- independent methods comparisons
- recovery of known additions
- calibration check standards
- blanks
- replicate analyses.
Inter-laboratory collaborative testing programs can also be used to assess the appropriateness of sample storage and preservation procedures. Statistically true values have confidence intervals associated with them, and sample measurements that lie within the confidence intervals will be considered to be accurate.
Analysis of certified reference materials and internal evaluation samples
Certified reference materials are materials of known concentrations that have a matrix similar to that of the sample being analysed. The accuracy of laboratory methods and procedures can be established by comparing the values for an analyte in the certified reference material against the results obtained by the laboratory for the same analyte. Results within the confidence limits specified for the certified reference material are deemed acceptable. Certified reference materials can be obtained from:
- European Commission Joint Research Centre (JRC) Reference Materials (Belgium)
- International Atomic Energy Agency (IAEA) (Austria)
- National Institute for Environmental Studies Certified Reference Materials (CRMs) (Japan)
- National Institute of Science and Technology (NIST) Standard Reference Materials (SRMs) (USA)
- National Research Council Canada Certified Reference Materials (Canada).
Internal evaluation samples is a general term for samples prepared by an outside source or by laboratory staff that have a known analyte concentration (e.g. certified reference materials). The acceptable range of measurement (recovery and precision) is determined for these samples, and analysts are expected to be within this range in all their analyses of these evaluation samples.
Proficiency testing programs (inter-laboratory comparisons)
Inter-laboratory comparison of unknown samples is used for testing instrument calibration and performance and the skills of the operator. Testing authorities frequently sponsor these programs.
Generally only a modest degree of sample preparation is required, probably to restrict the range of sources of variance between laboratories.
An individual laboratory compares its results against the consensus values generated by all the laboratories participating in the program, to assess the accuracy of its results and, hence, the laboratory procedures. It’s possible for the consensus values to be wrong, and there should be a known value from the authority conducting the proficiency program. Results within the confidence limits specified for the unknown samples are deemed acceptable.
Performance audits
During performance audits, unscheduled checks are made to detect deviations from standard operating procedures and protocols, and to initiate corrective action.
Independent methods comparison
The accuracy of analytical procedures can be checked by analysing duplicate samples by 2 or more independent methods. For the methods to be independent, they must be based on different principles of analysis.
For example, the determination of iron in water can be checked by physical or chemical principles, such as atomic absorption spectrometry (light absorption by atoms) vs anodic stripping voltametry (electrochemical reduction).
Bias in methods (interferences, insensitivity to chemical species) can cause 2 methods to give different results on duplicate samples. The average values obtained by the methods are compared using a Student’s t-test.
Recovery of known additions
By spiking a test sample with a known amount of analyte, it is possible to estimate the degree of recovery of the analyte and hence the accuracy of the method used. Spiking is one way of detecting loss of analyte. It is assumed that any interference or other effects that bias the method will affect the analyte spike and the analyte in the test sample in similar ways. Hence acceptable recoveries of the spike confirm the accuracy of the method.
This approach may be invalid if:
- chemical species that are added are different to the native chemical species in the sample and therefore undergo different processes and interferences (e.g. spiking of marine biological samples with AsO32– when native arsenic is present as arsenobetaine, C5H11AsO22–)
- interference is dependent on the relative concentrations of the analyte and interferent (addition of a spike will change this dependence and hence the magnitude of the interference)
- interference is constant regardless of the analyte concentration (recoveries can be quantitative but analysis of the native analyte may have large errors).
Calibration check standards
Standard curves (calibration curves) must be verified daily by analysing at least 1 standard within the linear calibration range. This ensures that the instrument is giving the correct response and reduces the likelihood of concentrations in samples being underestimated or overestimated.
Blanks
Blanks should be incorporated at every step of sample processing and analysis. However, only those blanks that have been exposed to the complete sequence of steps within the laboratory will routinely be analysed, unless contamination is detected in them. Blanks incorporated at intermediate steps are retained for diagnostic purposes only, and should be analysed when problems occur, to identify the specific source of contamination.
In principle, only field blanks need to be analysed in the first instance because they record the integrated effects of all steps. However, a laboratory will normally wish to test the quality of its internal procedures independently of those in the field, so laboratory procedural blanks will usually be included in a suite for analysis, in addition to field blanks.
Blanks cannot be used to detect loss of analyte. They are useful only to detect contamination. Blanks are particularly useful in detecting minor contamination, where the superimposition of a small additional signal on a sample of known concentration may not be evident in the statistical evaluation of analytical data. In other words, blanks are more sensitive to contamination.
If any blank measurement is further than 3 SDs from the mean, or if 2 out of 3 successive blanks measurements are further than 3 SDs from the mean, discontinue the analyses to identify and correct the problems.
Duplicate analyses
Duplicate analyses of samples are used to assess precision. At least 5% of samples should be analysed in duplicate.
QA/QC in biological analyses
For biological analyses, quality control procedures are designed to establish an acceptable standard of subsampling, sorting and identification.
Subsampling and sorting
For quality control of subsampling and sorting, a subsample equivalent in size to the original subsample should be sent to an independent group to check.
With macroinvertebrates, the data from the 2 subsamples are analysed to compare community composition and structure. The analysis compares the ratios of numbers of taxa and the Bray–Curtis dissimilarities in each subsample. After a sample has been sorted, the remainder is checked for macroinvertebrates that have been missed. Checking continues until more than 98% of the macroinvertebrates in the subsample have been consistently removed.
Identification
All organisms should be identified against taxonomic keys. If keys are not available, then preserved samples should be sent to other laboratories or museums that regularly identify similar samples/taxa.
Staff who identify biological specimens should be trained in the use of keys, and their proficiency should be tested before they are given responsibility for the analysis of samples.
For example, in the AUSRIVAS program, new staff identify organisms to family level in samples, and experienced staff check the same samples. The 2 lists of families are compared and discrepancies are discussed until the new staff understand their errors. The new staff continue to check samples but fewer samples are cross-checked by the experienced staff as the new staff improve. As many as 2 samples in 10 may be cross-checked in the early stages of training; later this drops to 2 samples in 50. New staff are considered proficient once their error rate at identification to family level is less than 10%.
For other taxa, and for macroinvertebrates where there is any uncertainty in the identification, museum taxonomic collections should be consulted to confirm the identities of the specimens. Where routine sample analysis is expected, the development of an internal laboratory voucher specimen collection is often the most efficient way to ensure continued accuracy of the identifications.
QA/QC in ecotoxicity testing
Important principles that are essential to enable sound ecotoxicological assessment to be achieved (Harris et al. 2014) include:
- completing essential aspects of experimental design and measurements
- demonstrating that the desired exposure conditions have been achieved
- choosing appropriate monitoring or control reference conditions, endpoints and analysis
- achieving an unbiased analysis of the results.
Essential aspects concern the baseline for the response endpoints (variability in responses for unexposed organisms), the use of appropriate exposure routes and concentrations (relevant to the environment being assessed) and the measurement and reporting of the exposure (conditions and concentrations of contaminant and non-contaminant stressors, such as ammonia).
Quality assurance procedures in ecotoxicity tests include criteria for test acceptability, appropriate positive and negative controls, use of reference toxicants, and water quality monitoring throughout the bioassays.
In ecotoxicity testing, any variability in the test organisms or their health is critical to the quality of the ecotoxicity results. For this reason, standard protocols specifying the life stage and health of an organism are essential. A comprehensive outline of toxicity testing procedures for sediments is given in Simpson & Batley (2016).
Test acceptability criteria
All ecotoxicity tests should have criteria for test acceptability. You should use statistical analyses to determine if toxicity has occurred and if the test acceptability criteria are met (ASTM 2013).
Basic statistical endpoints, such as percentage survival (mean ± SD) or the percentage of impairment (e.g. growth, reproduction, behaviour) should be calculated for each treatment and compared with the control and reference water or sediment results. These criteria are particularly important when the test is based on organisms collected in the field, which may vary in their response from season to season.
For example, in growth inhibition tests with microalgae, the growth rates of the control group of organisms must exceed a predefined daily doubling rate with less than 20% variability. Similarly, in acute tests with invertebrates and fish, at least 90% of the untreated control organisms must be alive after 96 hours. Fertilisation or reproduction tests usually specify an acceptable fertilisation rate, abnormality rate or number of offspring produced.
The criteria for determining if a test sediment is toxic usually specify a magnitude of effect (e.g. ≥ 20% lower survival or reproduction than the control or reference sediment) but will generally consider the past performance of the test (both inter- and intra-laboratory) in relation to the variability in responses that are typical of the type of sediments being tested.
If these criteria are not met, then the tests are usually determined to be invalid and will require repeating or explanations for the variance (e.g. from detection, through measurement and reporting, of unexpected non-contaminant stressors, such as ammonia).
Negative controls
All toxicity tests require the use of controls to compare the responses of the organisms in the presence or absence of toxicant. Negative controls can be uncontaminated seawater or freshwater used as diluent in the toxicity tests, with water quality characteristics similar to those of the test water.
For sediment tests, negative controls include uncontaminated sediments that have similar physical and chemical properties (e.g. particle sizes, organic carbon and sulfide contents) to those of the test sediment.
Many sediments ecotoxicity test programs may need multiple negative controls to cover the full range of properties of the test sediments (ranging from sandy to silty).
Reference toxicants
Reference toxicants or positive controls are used to ensure that the organism on which the toxicity test is based is responding to a known contaminant in a reproducible way. This is particularly important for field-collected organisms, which may vary in response to a toxicant depending on season, collection site, temperature and handling.
Reference toxicants are used to track changes in sensitivity of laboratory-reared or cultured organisms over time.
For water-only tests, usually either inorganic (e.g. copper, chromium) or organic (e.g. phenol, sodium dodecyl sulfate) reference toxicants are used and tested at a range of concentrations on a regular basis. Each toxicity test should include at least one concentration of reference toxicant as a positive control. Quality control charts are produced showing the mean response and variability over time. Refer to EC (1990) and USEPA (2002b) for guidance on the use of reference toxicant tests.
For sediments, the use of reference toxicants is complicated by the influence of sediment properties on bioavailability (Simpson & Kumar 2016). This is why reference toxicant tests for whole-sediment ecotoxicity tests are typically conducted as water only, short-term (e.g. 96-hour to 7-day static tests) and generally with exposure to a single concentration or dilution series of a single chemical, although spiked-sediment exposures may also be used.
Blanks
For water-only tests, appropriate field and process blanks should be included in each toxicity test if the sample has been manipulated before testing.
If freshwaters have to be salinity-adjusted with artificial sea salts before testing, sea salt controls should also be included in each test.
Solvent controls are essential for water-insoluble chemicals if they have to be dissolved in organic solvents to deliver them into the test system. It is important to test a solvent control with the same concentration of solvent in clean water as is found in the highest test concentration. The concentration of solvents or emulsifiers should never exceed 0.1 mg/L (OECD 1981).
For whole-sediment tests, the negative control prepared from sediment containing negligible contamination but similar physical and chemical properties plays the role of the blank.
Quality of ambient water
Throughout the toxicity tests, the quality of the organisms’ ambient water must be monitored to ensure that the toxicity measured is due to the contaminant or test sample alone and to provide information that can be used in test interpretation.
Measurements of alkalinity, hardness, pH, temperature and dissolved oxygen are the minimum parameters required for freshwaters and marine waters. For marine studies, salinity is also monitored throughout the test.
QA/QC for handling sediments
The principles of handling sediment samples are similar to those for water analyses and outlined in Simpson & Batley (2016). Sample integrity must be maintained and QA/QC are important, such as the need to maintain unchanged redox conditions (oxic vs anoxic) and the need to measure particle size, organic carbon and sulfide contents for interpretation of results. Refer to Simpson & Batley (2016) for advice on sediment handling and preparation.
Pore water sampling
Pore waters in sediments can be sampled by pressure filtration, centrifugation or in situ methods, such as pore water dialysis cells (peepers), gel samplers or sippers (Simpson & Batley 2016).
All operations should be conducted in an inert atmosphere. Filtration and centrifugation are the techniques most commonly used but their suitability will depend on the sediment grain size. Solvent displacement has also been successfully applied in centrifugation methods.
The use of sippers or direct withdrawal techniques is limited to sandy sediments with a large pore volume. For investigation of pore water depth profiles, the peeper and gel sampling methods offer the best option.
Sample storage
Sediment samples are typically either chilled or frozen immediately after collection, for storage and to minimise bacterial activity. Where total contaminant concentrations are of interest, either storage method is suitable.
Oven drying sediment samples at 110°C is another option. For most organic compounds and the more volatile metals (e.g. Hg, Cd, Se, As), oven drying is unacceptable, and air drying at room temperature or freeze drying is preferable, although even these may be a concern with very volatile organic compounds.
Special considerations are required where metal speciation is of concern or where pore waters are to be analysed. Since most sediments are anoxic, at least in part, oxidation of iron sulfides in particular will change the chemical forms of metals in the pore water and solid sediment phases. This oxidation can be minimised by freezing the sample and storing it in a sealed container, and by preferably carrying out such operations in an inert atmosphere (e.g. under a nitrogen gas blanket or in a glove box). Freeze-drying can be done in an oxygen-free environment, such as a glove box.
Freezing can rupture biological cells and release metals, which can significantly bias pore water results for some elements (e.g. Se).
Sieving samples
Sieving is the process used to divide sediment samples into fractions of different particle size.
Sediments are usually classified as:
- gravel (> 2 mm)
- sand (63 µm to < 2 mm)
- silt and clay (< 63 µm).
Sediments are usually sieved through a series of mesh sizes from 2 mm to 63 µm. Wet sieving is used for processing fine grain sediments. Sieving of dry material is used for the separation of coarser material. When comparing trace metal concentrations in sediments from different sampling sites, it is normal to analyse the < 63 µm fraction because it is this fraction that adsorbs most of the trace metals.
If organic contaminant concentrations in sediments are to be compared, then grain size is not important but concentrations should be expressed as a percentage of organic carbon content.
Homogenisation of samples
It is difficult to ensure the homogeneity of sediment samples being analysed because samples are notoriously heterogeneous with respect to particle size and contaminant distribution.
Thorough mixing of any wet or dried samples is required to improve homogeneity. For dry samples, grinding with a mortar and pestle is necessary. Commercially available rock grinders are used to reduce larger dried particles to less than 63 µm for analysis. Coning and quartering, rolling, mechanical mixing and splitting are used for homogenisation and selection of material for analysis (Simpson & Batley 2016).
Wet samples are used where it is feared that drying will alter the chemical form of the contaminants. For large wet sample volumes, homogenisation is especially difficult. It is more usual to use wet samples with smaller volumes (e.g. core sections). Here the wet samples can be homogenised by thorough mixing with a glass rod, then weighed out for analysis, with moisture determinations being carried out on separate aliquots of the sample.
Presentation of quality control data
Control charts are used to visualise and monitor the variability in quality control data (APHA 2012):
- Means charts are used to track changes in certified reference concentrations, known additions, calibration check standards and blanks. The charts are graphs of the mean ± SD or error over time (Figure 3) with a defined upper and lower control limit (normally 3 times the SD where 99.7% of the data should lie). Data that fall above or below these limits are unacceptable and corrective action must be taken. Normally, action is taken if data are trending towards these limits.
- Range charts are used to track differences between duplicate analyses based on the SD or relative SD. Limits are set, and when data fall above or below these limits, corrective action must be taken. Refer to APHA (2012) for a full explanation of the procedures for calculating ranges.
Figure3 An example of a control chart for mean values; UCL = upper control limit; LCL = lower control limit; UWL = upper warning limit; LWL = lower warning limit

The number of quality control samples will depend on 2 considerations:
- duration of acceptable SDs from the mean that can be tolerated
- likelihood of SDs occurring.
The first consideration will depend on the purpose for which the data are being collected, and the number of quality control samples will be set in consultation with the user of the data. The second consideration is usually based on past variability.
To be effective, control charts must be continually updated as data become available so trends can be established before control limits are reached.
As a minimum, the precision and accuracy of data must be stated when data are presented in reports. Precision, as SD or error, or relative SD or error, should be presented graphically across the range of values being measured (Figure 4a). Alternatively, the SD or similar should be given at discrete values that cover the range of measurements. The methods of establishing the accuracy of the data should be stated. Bias should be presented graphically as a function of the true value over the applicable range (Figure 4b). Alternatively, the bias at discrete values that cover the range of measurements should be given.
Figure 4 Graphical representation of (a) precision and (b) bias

Work health and safety
Legislative requirements
Work health and safety requirements for laboratory activities are provided in Safety in laboratories Part 2: Chemical aspects AS/NZS 2243.2:2006. This standard sets out the recommended procedures for safe working practices in laboratories and covers chemical aspects, microbiology, ionising radiators, non-ionising radiators, mechanical aspects, electrical aspects, fume cupboards and recirculating fume cupboards.
Practical guidance on the safety procedures and information needed for scientific work and safe laboratory practice is available in Haski et al. (1997).
Identification of hazards
The hazards or risks involved with laboratory work need to be identified and documented. The major issues are:
- exposure of staff to toxic or other hazardous substances
- placing staff in a position of potential physical danger.
Staff who are to conduct analyses should be physically and mentally able to carry out laboratory work.
Education about hazards
All staff must be appropriately trained as part of the formal risk minimisation strategy. Training includes:
- familiarisation with protocols (e.g. analysis procedures, safe handling procedures, disposal procedures, chain of custody considerations)
- use of laboratory equipment
- qualifications in handling chemicals
- familiarisation with safety procedures
- advanced qualifications in first aid.
Risk minimisation plans
Proper professional practice requires that risks be reduced as much as possible and that staff do not have to work in unsafe conditions.
Actions to reduce risks include:
- wearing appropriate clothing and footwear to protect against accidental chemical spills
- providing an appropriate first aid kit in close proximity to where analyses are being undertaken
- providing an eye bath and safety shower in the laboratory
- training laboratory staff in first aid
- staying in contact with help and never working alone.
At least 3 staff should work together and be in contact with someone who can raise an alarm. There should be written procedures describing how emergency services are to be contacted.
References
APHA 1998, Standard Methods for the Examination of Water and Wastewater, 20th Edition, American Public Health Association, American Water Works Association and Water Environment Federation, Washington.
APHA 2012, Standard Methods for the Examination of Water and Wastewater, 22nd Edition, American Public Health Association, American Water Works Association and Water Environment Federation, Washington.
AS 1990, Australian Standard AS3550.4-1990 Waters Part 4: Determination of Solids — Gravimetric Method, Standards Australia, Homebush.
ASTM 2013, Standard Practice for Statistical Analysis of Toxicity Tests Conducted Under ASTM Guidelines ASTM E1847 - 96(2013), Book of Standards Vol. 11.06, ASTM International, West Conshohocken.
DEC 2004, Approved Methods for the Sampling and Analysis of Water Pollutants in New South Wales, Department of Environment and Conservation, Sydney.
EC 1990, Guidance Document on Control of Toxicity Test Precision Using Reference Toxicants, Environment Canada Environmental Technology Centre, Ottawa.
EPA 1992, Sydney Deepwater Outfalls Environmental Monitoring Program: Pre-commissioning Phase Vol. 10 Contaminants in sediments, prepared by L. Gray, NSW Environment Protection Authority, Bankstown.
EPAV 2009, Sampling and Analysis of Waters, Wastewaters, Soils and Wastes, publication IWRG701, Environment Protection Authority Victoria, Melbourne.
Grasshoff K, Kremling K & Ehrhardt, M 2007, Methods of Seawater Analysis, 3rd Edition (Online), WileyVCH, Weinheim.
Harris CA, Scott AP, Johnson AC, Panter GH, Sheahan D, Roberts M & Sumpter JP 2014, Principles of sound ecotoxicology, Environmental Science & Technology 48(6): 3100–3111.
Haski R, Cardilini G & Bartolo WC 1997, Laboratory Safety Manual: An Essential Manual for Every Laboratory, CCH Australia Ltd.
OECD 1981, OECD Guidelines for the Testing of Chemicals, Organisation for Economic Cooperation and Development, Paris.
Parsons TR, Maita Y & Lalli C 1985, A Manual of Chemical and Biological Methods for Seawater Analysis, Pergamon.
RD Instruments 1989, Acoustic Doppler Current Profilers: Principles of Operation: A Practical Primer, P/N 951-6069-00, Teledyne RD Instruments, San Diego.
Simpson SL & Batley GE (eds) 2016, Sediment Quality Assessment: A Practical Handbook, CSIRO Publishing, Canberra.
Simpson SL & Kumar A 2016, Sediment ecotoxicology, in: Simpson SL & Batley GE (eds), Sediment Quality Assessment: A Practical Handbook, CSIRO Publishing, Canberra.
USEPA 1982, Handbook for Sampling and Sample Preservation of Water and Wastewater, United States Environmental Protection Agency Environmental Monitoring and Support Laboratory, Cincinatti.
USEPA 1983, Methods for Chemical Analysis of Water and Wastes, United States Environmental Protection Agency Environmental Monitoring and Support Laboratory, Cincinatti.
USEPA 1994, Methods for the Determination of Metals in Environmental Samples: Supplement 1, United States Environmental Protection Agency Environmental Monitoring and Support Laboratory, Cincinatti.
USEPA 1996, Method 1632: Determination of Inorganic Arsenic in Water by Hydride Generation Flame Atomic Absorption, United States Environmental Protection Agency, Washington DC.
USEPA 1999, Test Methods for Evaluating Solid Waste, Physical/Chemical Methods, Third Edition, Final Update III-A, United States Environmental Protection Agency, Washington DC.
USEPA 2002a, Method 1631, Revision E: Mercury in Water by Oxidation, Purge and Trap, and Cold Vapour Atomic Fluorescence Spectrometry, United States Environmental Protection Agency, Washington DC.
USEPA 2002b, Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms, 5th Edition, United States Environmental Protection Agency, Washington DC.








